
Prever mudanças conformacionais de proteínas continua sendo um desafio importante na biologia computacional e na inteligência artificial. Avanços na aprendizagem profunda, como o AlphaFold2, eliminaram o objetivo de prever estruturas estáticas, mas não abordam as mudanças dinâmicas pelas quais muitas proteínas passam para desempenhar suas funções biológicas. Essas mudanças são importantes para a compreensão de uma ampla gama de processos biológicos, desde a atividade enzimática até a transdução de sinal. No entanto, a falta de dados estruturais para as regiões centrais dificulta a previsão destas mudanças. Além disso, os modelos existentes sofrem com elevadas barreiras de fluxo livre nas regiões de transição, tornando as previsões precisas ainda mais desafiadoras. Catalítico para o desenvolvimento em muitos campos, incluindo o desenvolvimento de medicamentos, a biologia sintética e a investigação de doenças, irá
Os modelos existentes para descrever mudanças conformacionais de proteínas incluem análise de modo normal de redes elásticas e modelos híbridos que combinam redes elásticas com simulações de dinâmica molecular. Esses métodos são adequados para movimentos conformacionais simples, mas não têm resolução para dar conta das mudanças complexas e multidimensionais encontradas em proteínas grandes. Recentemente, métodos de aprendizagem profunda, como codificadores automáticos, geradores de Boltzmann e modelos de difusão, foram desenvolvidos para projetar estruturas proteicas em espaços ocultos de baixa dimensão. No entanto, estes modelos baseiam-se num caminho linear entre dois estados, o que não é aplicável a mudanças complexas e não lineares, como a mudança de dobragem. Mais importante ainda, as altas demandas de dados e a baixa eficiência dos dados, além do custo computacional que proíbe muitas aplicações em tempo real, tornam esses métodos insatisfatórios.
Os autores elaboram uma nova estratégia de aprendizagem profunda usando amostras biofísicas elevadas para evitar a falta de dados relacionados à modificação de proteínas. Simulações de dinâmica molecular foram combinadas com métodos de amostragem aprimorados para gerar uma biblioteca de 2.635 proteínas com dois estados determinados experimentalmente. Este conjunto de dados usa um modelo abrangente de aprendizado profundo chamado PATHpre, que prevê caminhos estruturais que resultam em mutações conformacionais com alta precisão. Em particular, o desenvolvimento do módulo HESpre no PATHpre preocupa-se com o desempenho preditivo do estado de alta energia na via de transição. O modelo proposto não faz uma suposição de espaço latente direto que possa ser objeto de críticas. Mostra a disponibilidade geral de proteínas em diferentes espécies. Isso significaria uma grande contribuição que aborda a simulação do comportamento dinâmico em sistemas complexos, utilizando medição e eficiência de dados em nível metodológico.
No método PATHpre, as matrizes de distância na matriz de dois estados de conformação são usadas pela previsão da rede neural de convolução para encontrar o estado com a energia mais alta entre esses estados de conformação; é aqui que o HESpre se concentra: apenas as conexões especiais ou únicas que ele encontra ocorrem com a maior potência do par residual em cada rota com base na matriz de distância aos pares, ele mede a estrutura e a quebra da rota percorrida e a matriz de conexão geral é estabelecido. Ele contém quatro classes de proteínas MS isoladas que exibem movimento interdomínio e intradomínio, localização e mudanças globais em suas propriedades conformacionais. A validação cruzada para várias proteínas foi realizada para o modelo, que alcançou fortes correlações de Pearson e baixos erros médios absolutos para todas as medidas; portanto, é altamente variável em todas as fases estruturais. O bom desempenho geralmente estabelece o desempenho geral do modelo em proteínas de comprimento de sequência e complexidade estrutural variados.
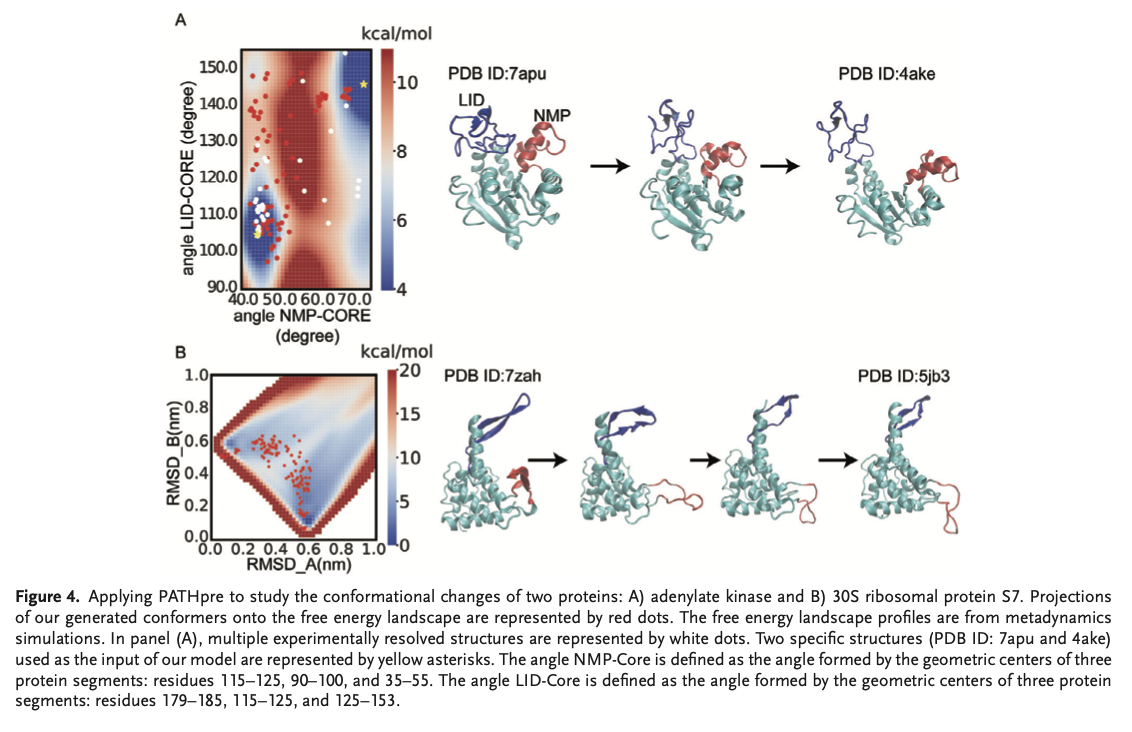
PATHpre é altamente preciso na previsão de caminhos mutacionais de proteínas, mostrando fortes correlações com dados experimentais e simulados em uma variedade de proteínas. Os experimentos também mostraram que o PATHpre captura de forma robusta mudanças conformacionais simples a complexas e é compatível com vários comprimentos de sequência e complexidades estruturais. É importante ressaltar que ele previu com precisão os modos de transição de proteínas individuais, como a adenilato quinase e a proteína ribossômica 30S S7, combinando a sonda de energia livre e teve um desempenho melhor do que os métodos híbridos convencionais em situações desafiadoras. As previsões do PATHpre foram alinhadas com estruturas conhecidas, e seu mapeamento de regiões intermediárias positivas para proteínas variáveis confirmou sua ampla aplicabilidade e confiabilidade para capturar uma ampla gama de alterações conformacionais de proteínas.
Este trabalho marca um avanço significativo na modelagem de proteínas orientada por IA, fornecendo um método confiável e eficiente em termos de dados para prever mudanças conformacionais de proteínas. A integração de uma grande amostra biofísica e aprendizagem profunda no PATHpre aborda o forte desafio dos dados limitados e captura mudanças não específicas na diversidade de proteínas. Este modelo generalizável será a base para o uso altamente desenvolvido de aplicações de IA em biologia computacional, estabelecendo assim uma ferramenta poderosa para investigar o comportamento dinâmico de proteínas em diferentes contextos – desde a descoberta de medicamentos até a biologia sintética.
Confira Papel. Todo o crédito deste estudo vai para os pesquisadores deste projeto. Além disso, não se esqueça de nos seguir Twitter e junte-se ao nosso Estação telefônica de novo LinkedIn Gracima. Se você gosta do nosso trabalho, você vai gostar do nosso jornal.. Não se esqueça de participar do nosso SubReddit de 55k + ML.
[Sponsorship Opportunity with us] Promova sua pesquisa/produto/webinar para mais de 1 milhão de leitores mensais e mais de 500 mil membros da comunidade
Aswin AK é consultor da MarkTechPost. Ele está cursando seu diploma duplo no Instituto Indiano de Tecnologia, Kharagpur. Ele é apaixonado por ciência de dados e aprendizado de máquina, o que traz consigo uma sólida formação acadêmica e experiência prática na solução de desafios de domínio da vida real.
Ouça nossos podcasts e vídeos de pesquisa de IA mais recentes aqui ➡️


