
A geração de estruturas proteicas atômicas é um grande desafio na síntese protéica de novo. Os modelos da geração atual são altamente avançados na geração de backbone, mas permanecem difíceis de resolver com precisão atômica porque a identidade única dos aminoácidos está incorporada no posicionamento contínuo dos átomos no espaço 3D. Este problema é particularmente importante na concepção de proteínas funcionais, incluindo enzimas e ligantes moleculares, uma vez que pequenas imprecisões à escala atómica podem impedir aplicações práticas. Adotar uma nova estratégia que possa abordar eficazmente estes dois aspectos, preservando simultaneamente a precisão e a eficiência computacional, é essencial para superar este desafio.
Modelos atuais como RFDiffusion e Chroma concentram-se principalmente em configurações de backbone e oferecem resolução atomística limitada. Extensões como RFDiffusion-AA e LigandMPNN tentam capturar a complexidade do nível atômico, mas não podem representar totalmente as configurações de todos os átomos. Métodos baseados em superposição, como Protpardelle e Pallatom, tentam aproximar estruturas atômicas, mas sofrem com altos custos computacionais e desafios no tratamento de interações contínuas. Além disso, estes métodos lutam para conseguir um compromisso entre sequência estrutural e diversidade, o que os torna menos úteis para aplicações práticas na concepção de proteínas autênticas.
Pesquisadores da UC Berkeley e UCSF estão apresentando o ProteinZen, uma estrutura de produção em dois estágios que combina fluxo paralelo de quadros de fundo com modelagem de espaço latente para obter uma produção de proteínas atomicamente precisa. No primeiro estágio, o ProteinZen constrói estruturas de backbone de proteínas dentro do espaço SE(3) enquanto gera simultaneamente representações latentes de cada resíduo usando métodos de simulação de fluxo. Esta extração básica evita, portanto, uma correlação direta entre a posição do átomo e a identidade do aminoácido, o que facilita muito o processo de produção. Nesta próxima etapa, o VAE híbrido com MLM traduz as representações latentes em estruturas de nível atômico, prevê ângulos de torção da cadeia lateral e identidades de sequência. A inclusão da perda de passe melhora o alinhamento das estruturas geradas com as estruturas atômicas originais, garantindo maior precisão e consistência. Esta nova estrutura aborda as limitações dos métodos existentes, alcançando precisão em nível atômico sem sacrificar a versatilidade e a eficiência computacional.
ProteinZen usa fluxo paralelo SE(3) para gerar o backbone e o fluxo euclidiano paralelo para recursos latentes, minimizando a perda de rotação, translação e prevendo a representação latente. Um autoencoder híbrido VAE-MLM integra informações atômicas em variáveis latentes e as separa em sua ordem e configuração atômica. A estrutura do modelo inclui codificação Tensor-Field Networks (TFN) e camadas IPMP otimizadas para codificação, garantindo equivalência SE(3) e eficiência computacional. O treinamento foi realizado no conjunto de dados AFDB512, que foi cuidadosamente construído combinando monômeros compostos PDB e representantes do banco de dados AlphaFold contendo proteínas com até 512 resíduos. O treinamento deste modelo utiliza uma combinação de dados reais e artificiais para melhorar a generalização.
ProteinZen atinge 46% de identidade de sequência estrutural (SSC), superando os modelos existentes, mantendo alta diversidade estrutural e de sequência. Ele equilibra bem precisão e inovação, produzindo estruturas proteicas diferentes, mas únicas, com precisão competitiva. A análise de desempenho mostra que o ProteinZen é eficiente para sequenciamento de pequenas proteínas, ao mesmo tempo que se mostra promissor no desenvolvimento para modelagem de longo alcance. As amostras sintetizadas variaram de uma variedade de estruturas secundárias, com uma fraca tendência para alfa-hélices. A análise estrutural confirma que a maioria das proteínas produzidas está em conformidade com lacunas conhecidas, ao mesmo tempo que mostra adaptação a novas dobras. Os resultados mostram que o ProteinZen pode produzir estruturas de proteínas atômicas altamente precisas e diversas, marcando assim uma melhoria significativa em relação aos métodos de produção existentes.
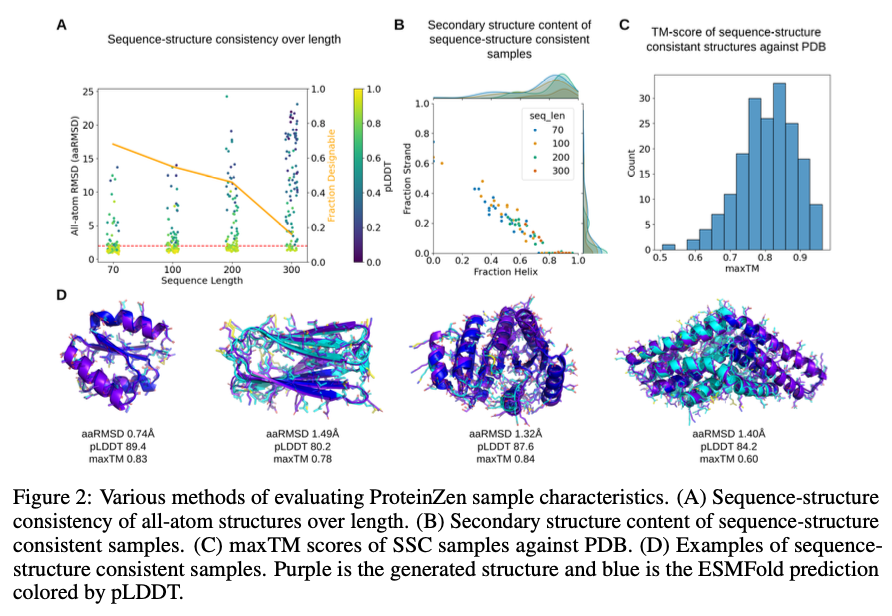
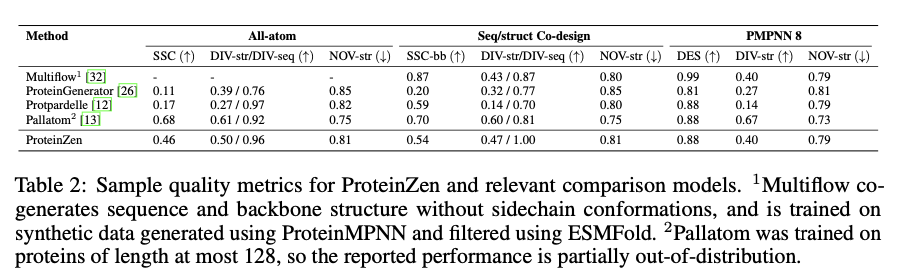
Concluindo, ProteinZen apresenta um novo método para modelagem de proteínas de átomos inteiros, combinando síntese de backbone paralelo SE (3) e simulações de fluxo latente para reconstruir estruturas atômicas. Ao distinguir as diferentes identidades dos aminoácidos e o posicionamento contínuo dos átomos, o método atinge precisão no nível atômico, preservando ao mesmo tempo a diversidade e a eficiência computacional. Com 46% de sequência estrutural consistente e evidência de variação estrutural, o ProteinZen estabelece um novo padrão para modelagem de proteínas sintéticas. O trabalho futuro incluirá o desenvolvimento de modelagem estrutural de longo alcance, o refinamento da interface entre o espaço latente e o decodificador e a avaliação de funções de design condicional de proteínas. Este desenvolvimento representa um avanço significativo em direção ao design preciso, eficiente e realista de átomos de proteínas inteiras.
Confira eu Papel. Todo o crédito deste estudo vai para os pesquisadores deste projeto. Além disso, não se esqueça de nos seguir Twitter e junte-se ao nosso Estação telefônica de novo LinkedIn Gracima. Não se esqueça de participar do nosso SubReddit de 60k + ML.
🚨 Tendências: LG AI Research Release EXAONE 3.5: Modelos de três níveis de IA bilíngue de código aberto oferecem seguimento de comando incomparável e insights profundos de conteúdo Liderança global em excelência em IA generativa….
Aswin AK é consultor da MarkTechPost. Ele está cursando seu diploma duplo no Instituto Indiano de Tecnologia, Kharagpur. Ele é apaixonado por ciência de dados e aprendizado de máquina, o que traz consigo uma sólida formação acadêmica e experiência prática na solução de desafios de domínio da vida real.
🧵🧵 [Download] Avaliação do relatório de trauma do modelo de linguagem principal (estendido)


